.png)
Introducing Axis
October 8, 2025
Highlights
- Axis outperforms Google DeepMind’s AlphaGenome at predicting regulatory element activity by 6.7% on average.
- High-affinity prompts drive up to 9x enrichment of targeted transcription factor motifs versus low-affinity prompts.
- Training Axis to generate DNA sequences improved its ability to predict properties of existing sequences.
- Multifunctional models like Axis pave the path to safer and more effective gene therapies through regulatory element mediated control of gene expression.
Today, we are announcing Axis, the first AI model that generates regulatory DNA elements and also predicts their function. Gene and cell therapies have the promise of being lifelong cures that offer significant improvements to quality of life over common standard-of-care treatments. However, their potential remains unrealized due to poor efficacy, immune responses and off-tissue effects. AI-guided development can enable us to overcome these risks making them safer and more effective.
Current DNA models are limited in their utility and capabilities. They are restricted to natively perform either DNA-to-DNA, DNA-to-function or function-to-DNA tasks. Axis is the first to unify all three capabilities. This multifunctional design exposes the model to different dynamics within the same domain leading to richer representations of the rules that govern regulatory DNA. On benchmarks, Axis outperforms Google DeepMind’s AlphaGenome at predicting regulatory element binding activity by 6.7% on average. Additionally, when prompted with “high” binding affinity, regulatory sequences generated by Axis show up to 9x enrichment of targeted transcription factor (TF) binding sites when compared to “low” binding affinity prompts.
Unified and multifunctional models are the scalable path to reliable and capable AI systems. We built Axis as the first step to enable the therapeutic use of AI to optimize the safety, specificity, and effectiveness of gene and cell therapies.
The Promise & Perils of Gene Therapy
Gene and cell therapies are already changing lives: Luxturna has produced durable functional vision gains in RPE65-mediated retinal disease while Zolgensma has improved survival and motor function in infants with Spinal Muscular Atrophy (SMA). In oncology, CD19- and BCMA-directed CAR-T therapies have delivered deep and sometimes long-lasting remissions in otherwise refractory B-cell malignancies.
Despite these advances, the field still faces three persistent challenges: limited efficacy, immune/toxicity events, and off-tissue activity. In CAR-T cells specifically, high CAR surface density can drive tonic signaling and T-cell exhaustion, undermining persistence and antitumor activity. In AAV treatments, ectopic or overly strong expression increases antigen burden and presentation raising immunogenicity and tolerability risks, while under-expression can reduce potency.
This is exactly where promoter and enhancer optimization can help. In CAR-T cells, moving from very strong constitutive backbones to milder promoters (e.g., EFS/MND-class) lowers CAR density, reduces tonic signaling/cytokines, and preserves tumor killing capabilities, creating a direct way to rebalance efficacy and safety. In AAV gene therapy, tissue-specific promoters focus expression in target organs and limit off-tissue exposure; for Duchenne, the MHCK7 muscle-biased promoter with cardiac enhancement concentrates micro-dystrophin in skeletal and cardiac muscle (including the diaphragm), improving functional readouts while reducing unnecessary exposure elsewhere.
Gene Regulation & Mechanical Control
An epigenetic code governs the accessibility of DNA, allowing proteins called transcription factors to bind to specific sites. These sites are short nucleotide sequences (~8-10 bp long) and are called “motifs”. When TFs bind the motifs they identify, they switch on different genes within the DNA giving the cell its identity and function. The availability of the TFs differs from cell type to cell type. For example, REST is a TF that inhibits cells from making neuronal proteins and so it is expressed mainly in non-neuronal cells.
AI-designed regulatory sequences can enable precise control: TF-binding prompts let you condition sequence generation on the presence of transcription factors in your target cell state and type, biasing gene expression where those factors are active and reducing exposure elsewhere.
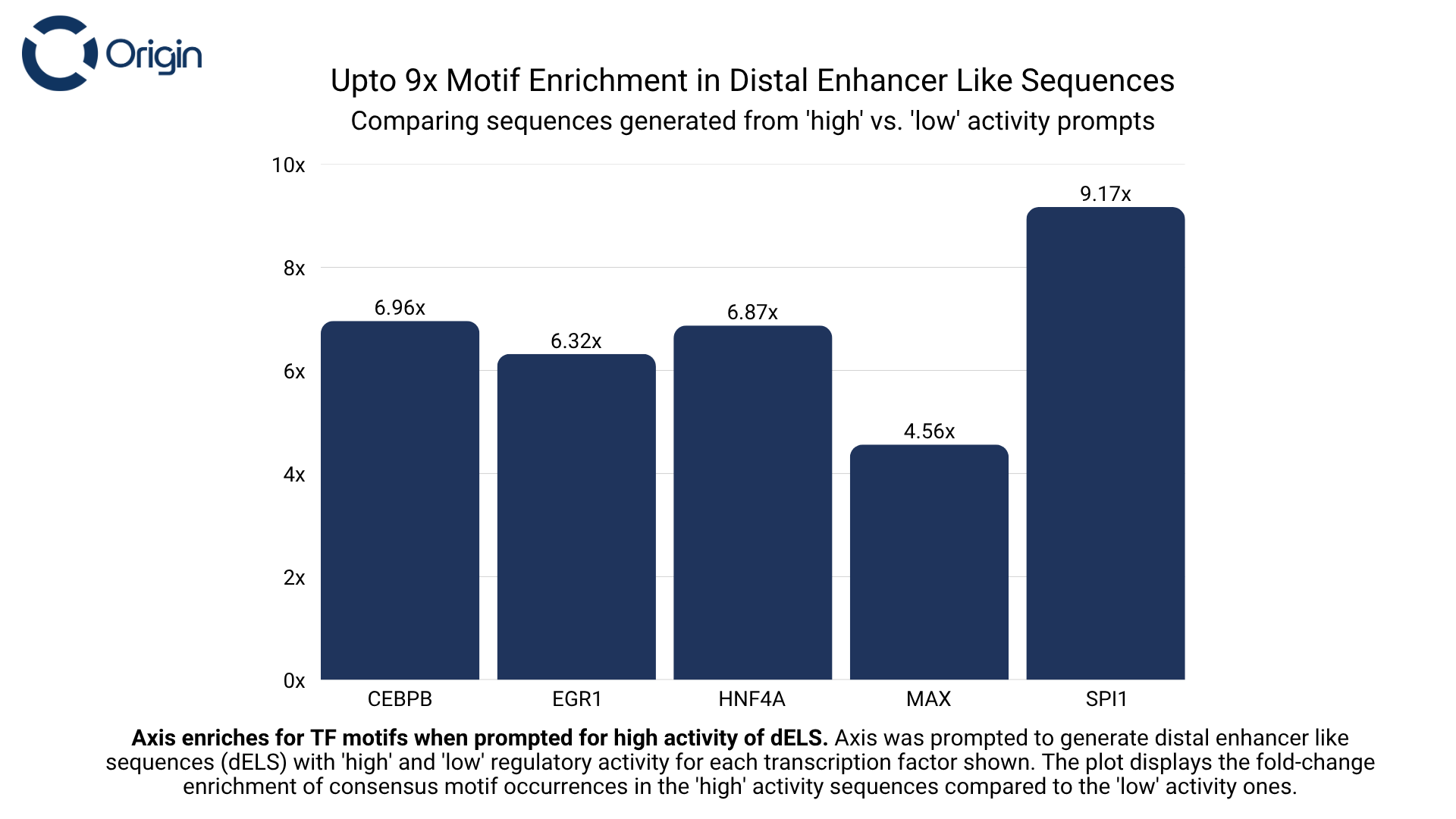
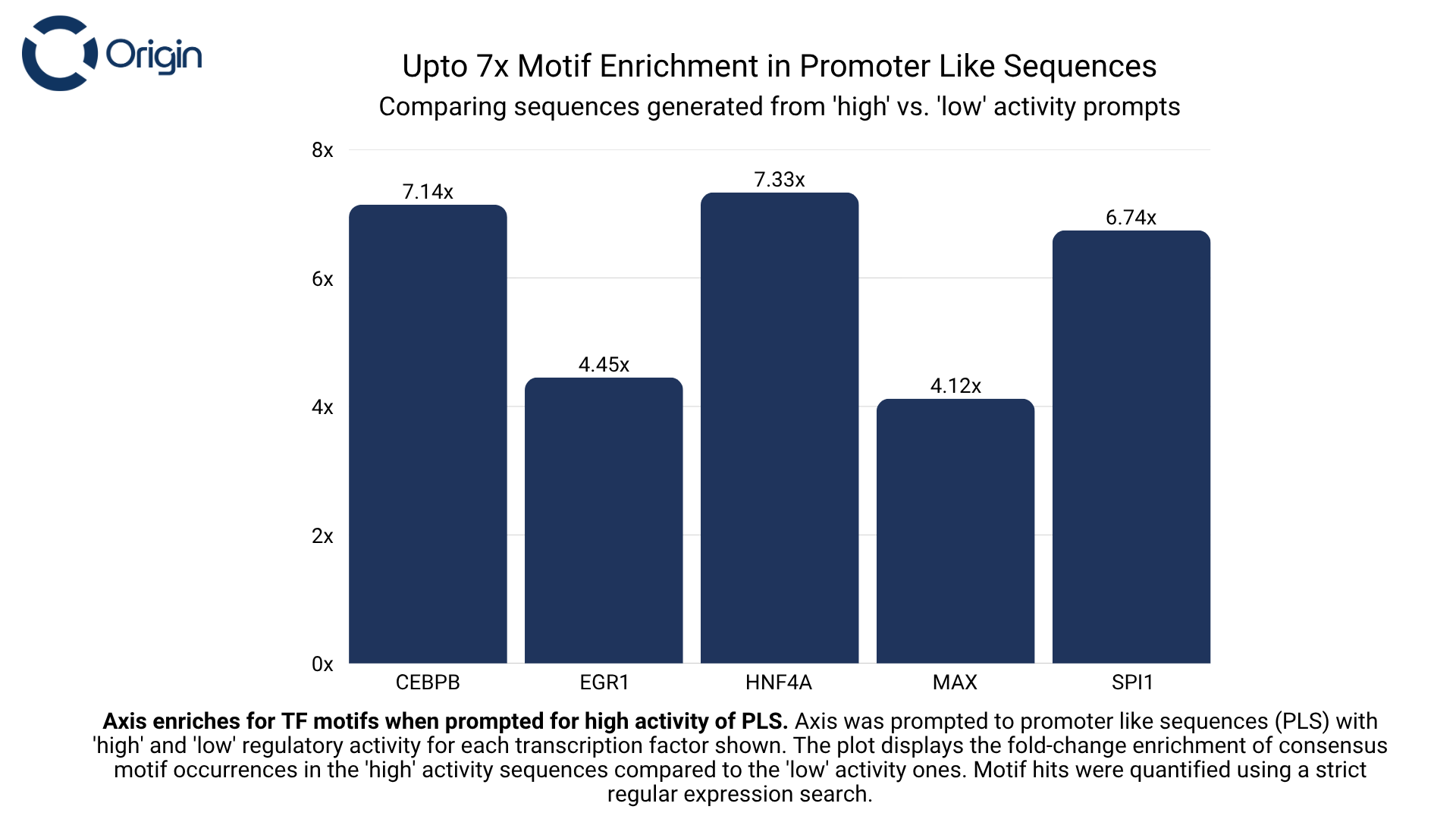
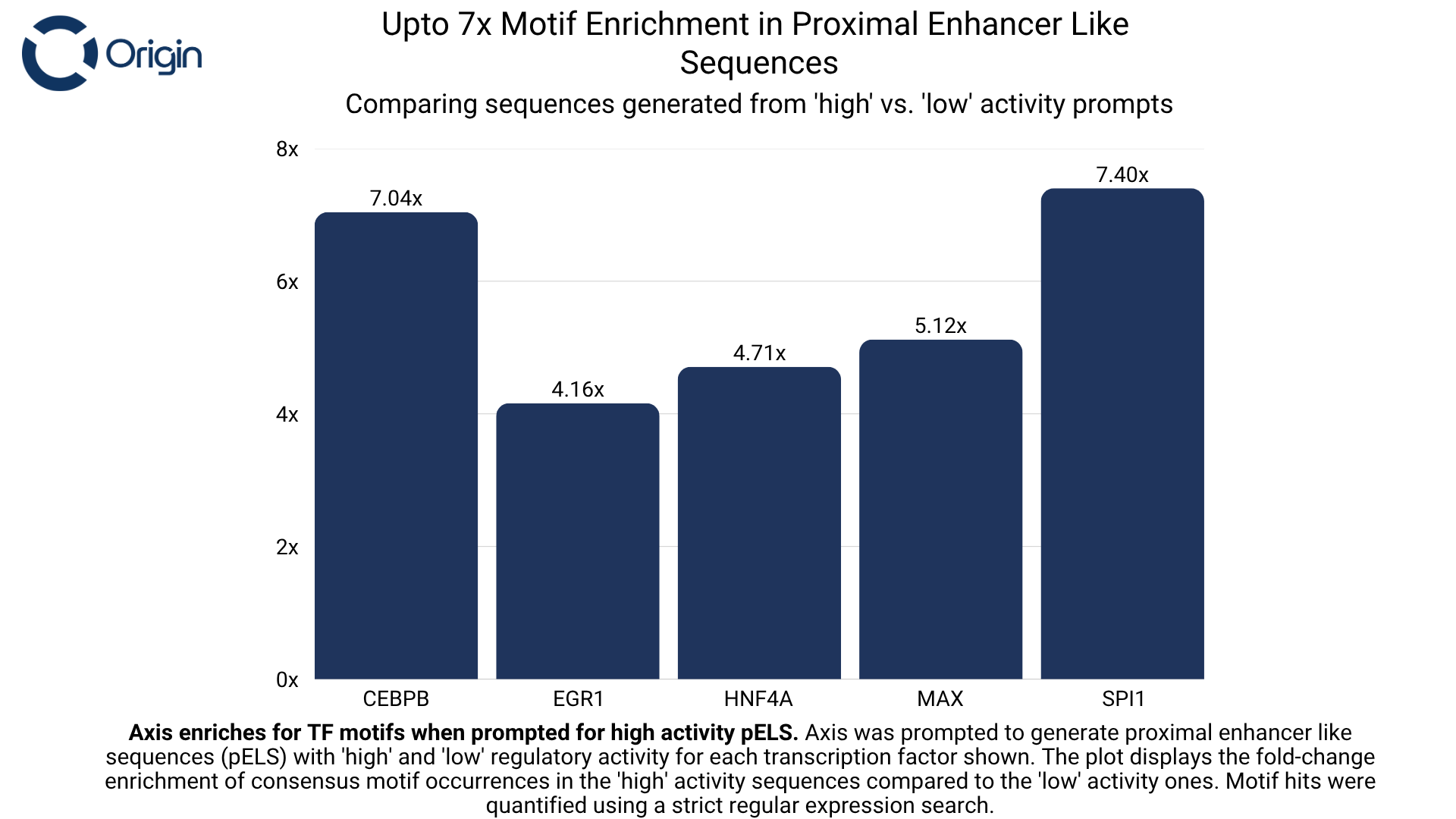
For every transcription factor that Axis was trained on, we prompted it to generate sequences with the intended binding of that transcription factor (Set A), and then asked it to generate sequences without explicit prompting of that same transcription factor (Set B). We observed that sequences from Set A had up to 9x more occurrences of the protein’s consensus motif compared to Set B. This motif enrichment shows that the model identifies the distinct role of these motifs and is also capable of following instructions.
The above evaluation was done separately on three regulatory types (see figures below):
- Distal Enhancer like Sequences (dELS): Usually found >2,000 bp away from a gene’s transcription start site (TSS). They’re involved in long-range control and activate target genes via enhancer-promoter contact.
- Proximal Enhancer like Sequences (pELS): Located within ~2,000 bp from a gene’s TSS. They modulate the nearby promoter and its responsiveness to signal.
- Promoter like Sequences (PLS): Found within 200 bp of a gene’s TSS. Involved in recruiting RNA Polymerase II to start gene transcription.
Distal Enhancer Sequences
Promoter Sequences
Proximal Enhancer Sequences
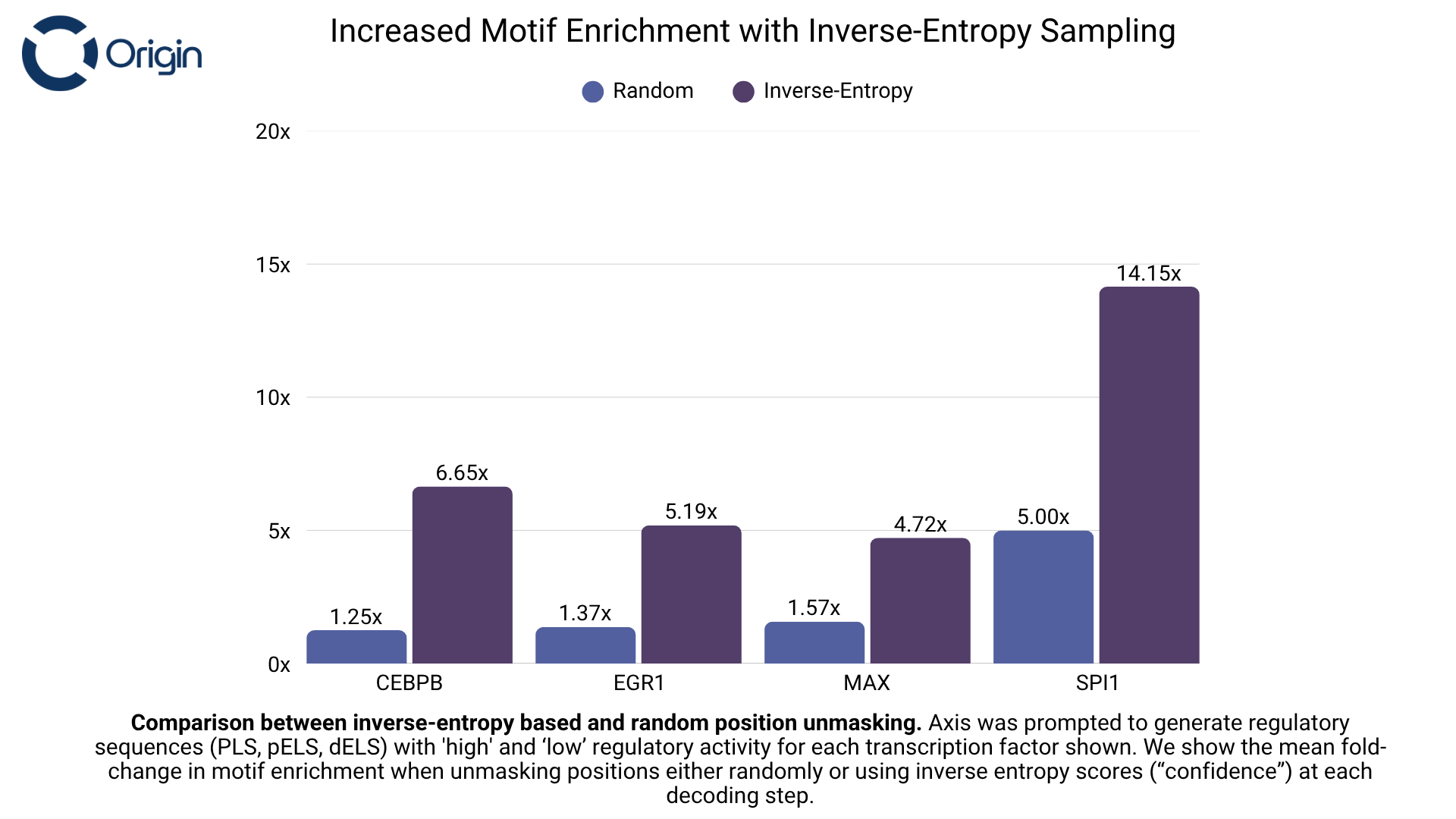
We used an inverse-entropy (“confidence-based”) sampling strategy to decide which positions to generate at each decoding step. Given the bidirectional nature of DNA and Axis’ learned understanding of motif spacing, placement, and orientation, we hypothesized that allowing the model to iteratively generate its most confident positions—conditioned on prior steps—would yield more biologically coherent sequences. To test this, we compared entropy-based sampling to a baseline that randomly selected which positions to generate (while still sampling nucleotides from the model’s logits). Our entropy-based approach led to up to a 5x increase in motif enrichment.
Exploring Larger Design Spaces
Generative models that learn the rules of transcription factor binding open up new ways to control and program gene therapies. Natural genomes only explore a fraction of the possible combinations of TF motifs, but synthetic design can uncover novel interactions and explore this design space. By capturing these rules, the model can generate enhancers and promoters that encode circuit-like logic - starting from single TF binding site effects and extending to combinatorial interactions between multiple TF motifs for greater specificity and complexity.
As we expand to joint conditioning with other modalities like expression activity, we expect significant increases in programmability. Here, we hope to convey the intuition of the kinds of programmability the model can utilize using analogies to circuit components. Recent work by the Centre for Genomic Regulation (CRG) demonstrates TF motif interactions that inspired the circuit interpretations shown below.
Single-TF motif circuit components
These are control circuits that can arise from the presence of binding sites of single transcription factors on a regulatory sequence (e.g., enhancers).
Switch
Enhancer sequences containing motifs for certain single transcription factors show switch-like behavior: they are active in certain cell states but silent or repressed in others. This demonstrates that even a single motif can encode selective control, providing cell-state-specific activity rather than a universal effect. Intuitively, it's similar to a switch that turns on only in particular rooms, highlighting how basic motif designs can already achieve targeted expression.
Dial
For some TFs that act primarily as activators (or primarily as repressors), increasing site number / strength tends to monotonically increase (or decrease) reporter activity. It's a straightforward, dosage-like control, with the response slope varying by cell state.
Several TFs exhibit a non-monotonic relationship: intermediate occupancy maximizes activation, while too many strong sites reduce activity or flip to repression. A single dial can therefore push one cell state above an activation threshold while pushing another below it.
Pairwise-TF motif circuit components
These are more complex control circuits that can arise from combinations of TF motif pairs on regulatory sequences.
XOR-like behavior
Some motif pairs show XOR-like behavior: each factor alone can activate in a given cell state, but when both are present together the enhancer is repressed.
Slider
For certain TF pairs, the ratio of their binding sites determines whether the enhancer activates or represses. This makes enhancers act like sliders that respond to relative abundance rather than absolute levels.
About the Model & Data
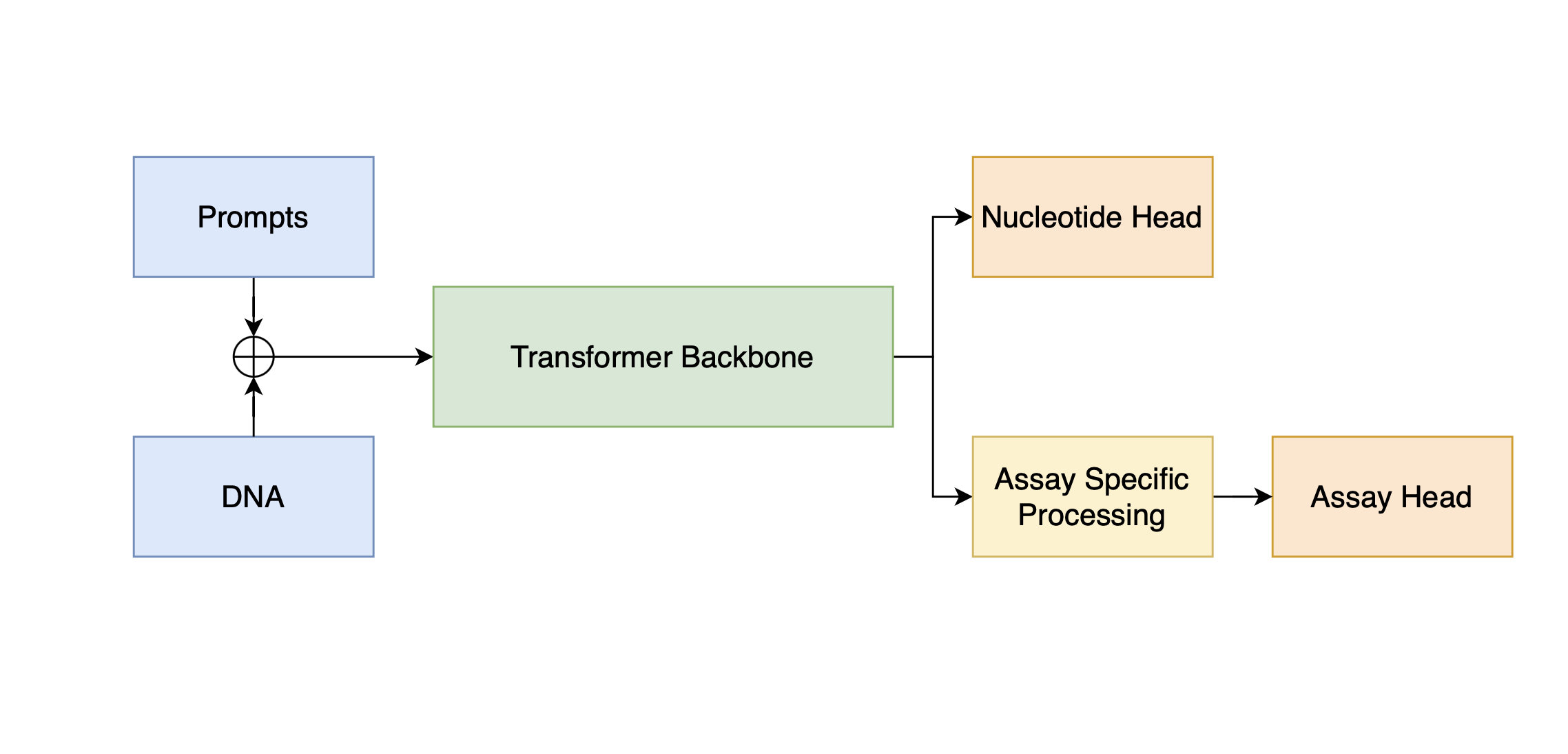
Our model architecture employs a shared Transformer backbone for the DNA generation and assay prediction tasks. Nucleotide and assay heads diverge after the final Transformer layer with some additional processing specific to the assay prediction in-between. Cell type and the prompts (regulatory type such as promoter + transcription factor proteins) are tokenized and appended to the input DNA sequence. The final loss is a linear combination of the cross-entropy for nucleotide prediction with a factorized Poisson and multinomial loss to capture the profile and intensity of the regulatory region.
The source of the training data is ENCODE. We use intervals of independent cis-regulatory elements (CREs) from the ENCODE V4 Registry found on the SCREEN page. CREs are ranked by their mean activity and we derive a z-score ranking for every biosample and assay combination. CREs are then split into train and test scaffolds and we verify that there is no overlap of genomic coordinates in order to ensure zero contamination in the data used for benchmarking. Our data processing pipeline of the actual experimental files is identical to what is done and reported in the AlphaGenome paper.
Benchmarks
Chromatin accessibility
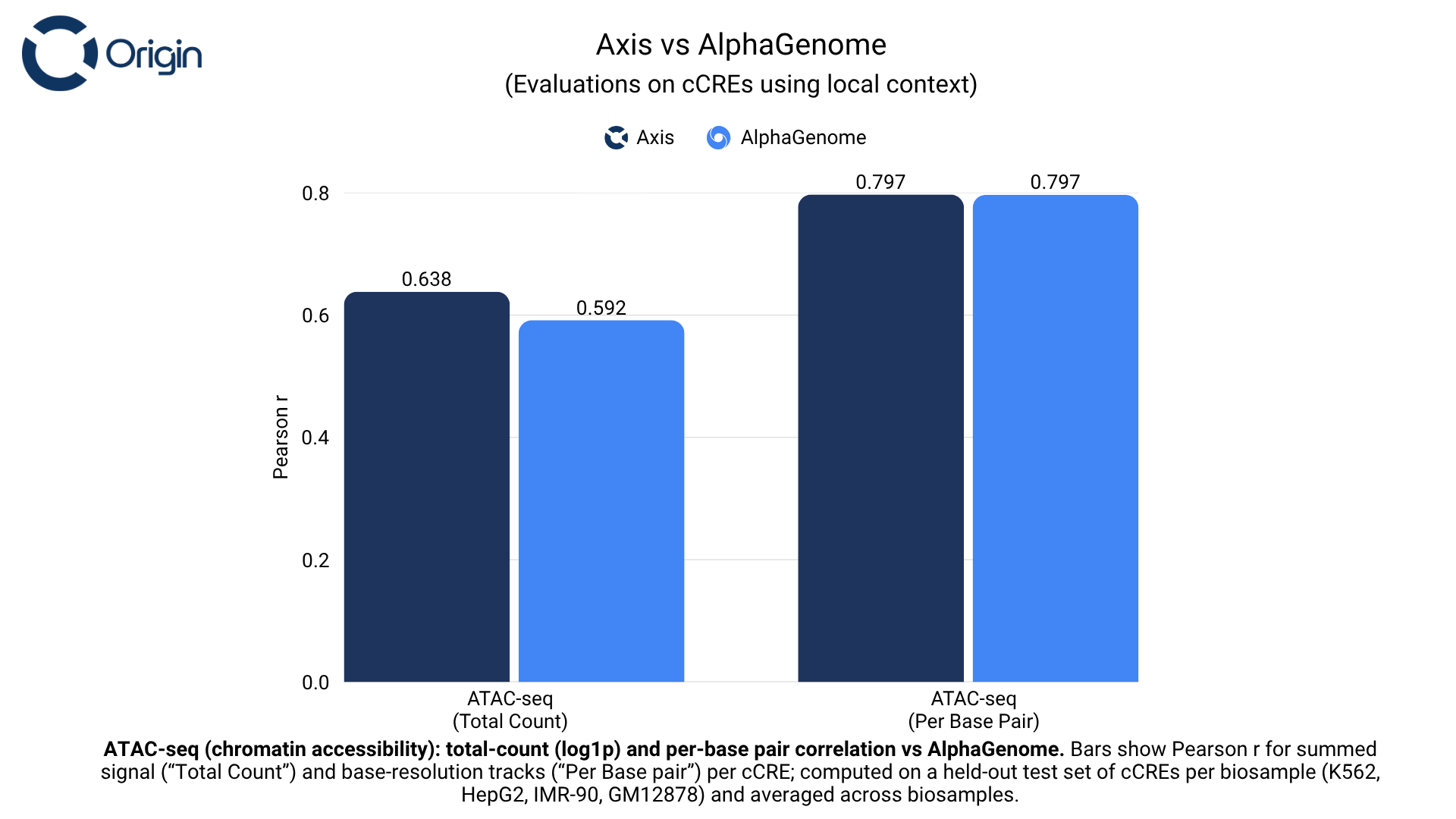
Chromatin accessibility measures open sequences across the genome, highlighting regulatory regions that are accessible for TF binding.
Lineage specification and maintenance factors
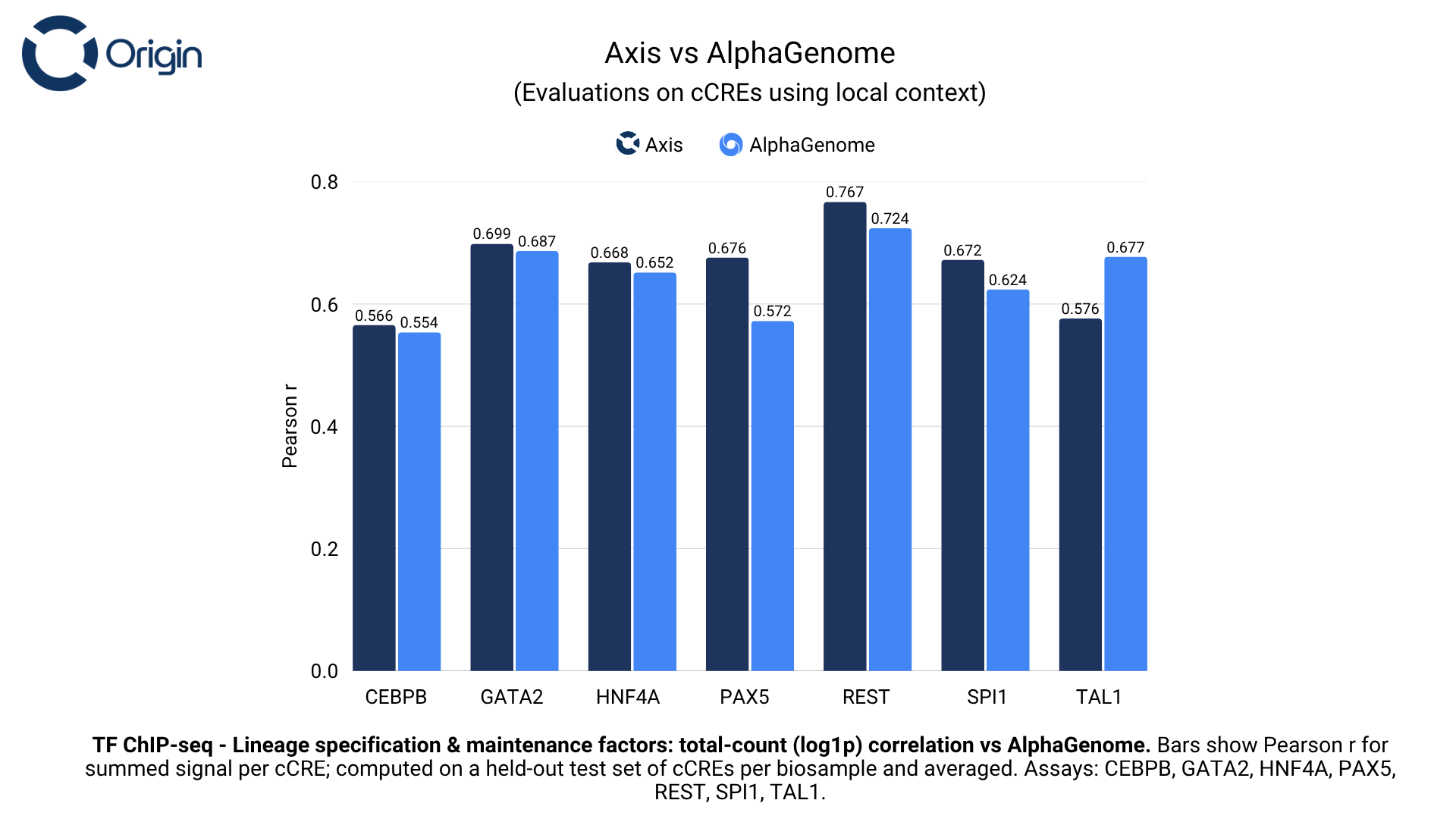
Lineage specification and maintenance transcription factors control the activation or repression of lineage-specific transcriptional programs to maintain cell identity and function.
Signal response factors
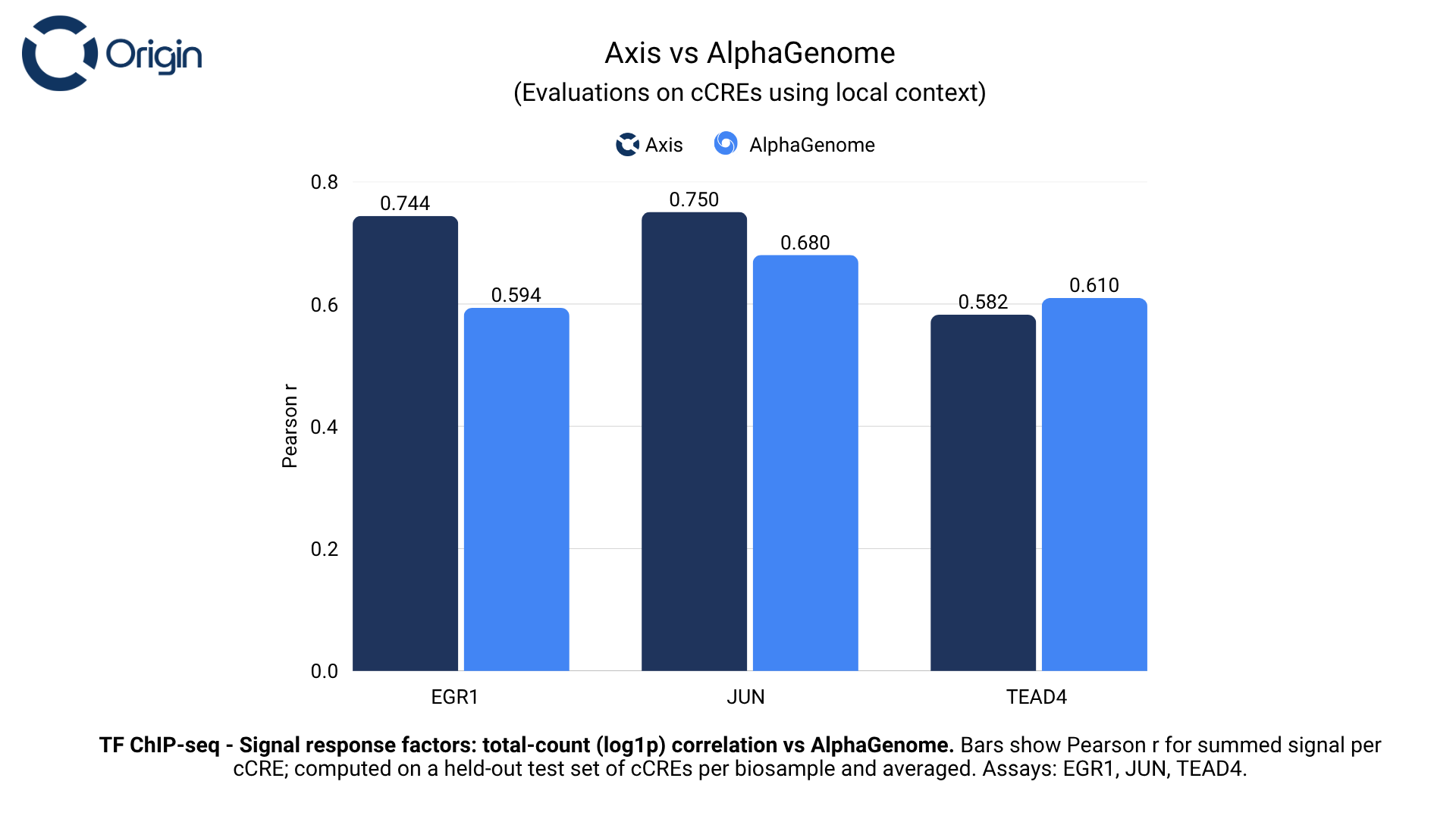
Signal response transcription factors act as sensors and responders, translating external stimuli into gene expression programs.
Chromatin architecture and growth control factors
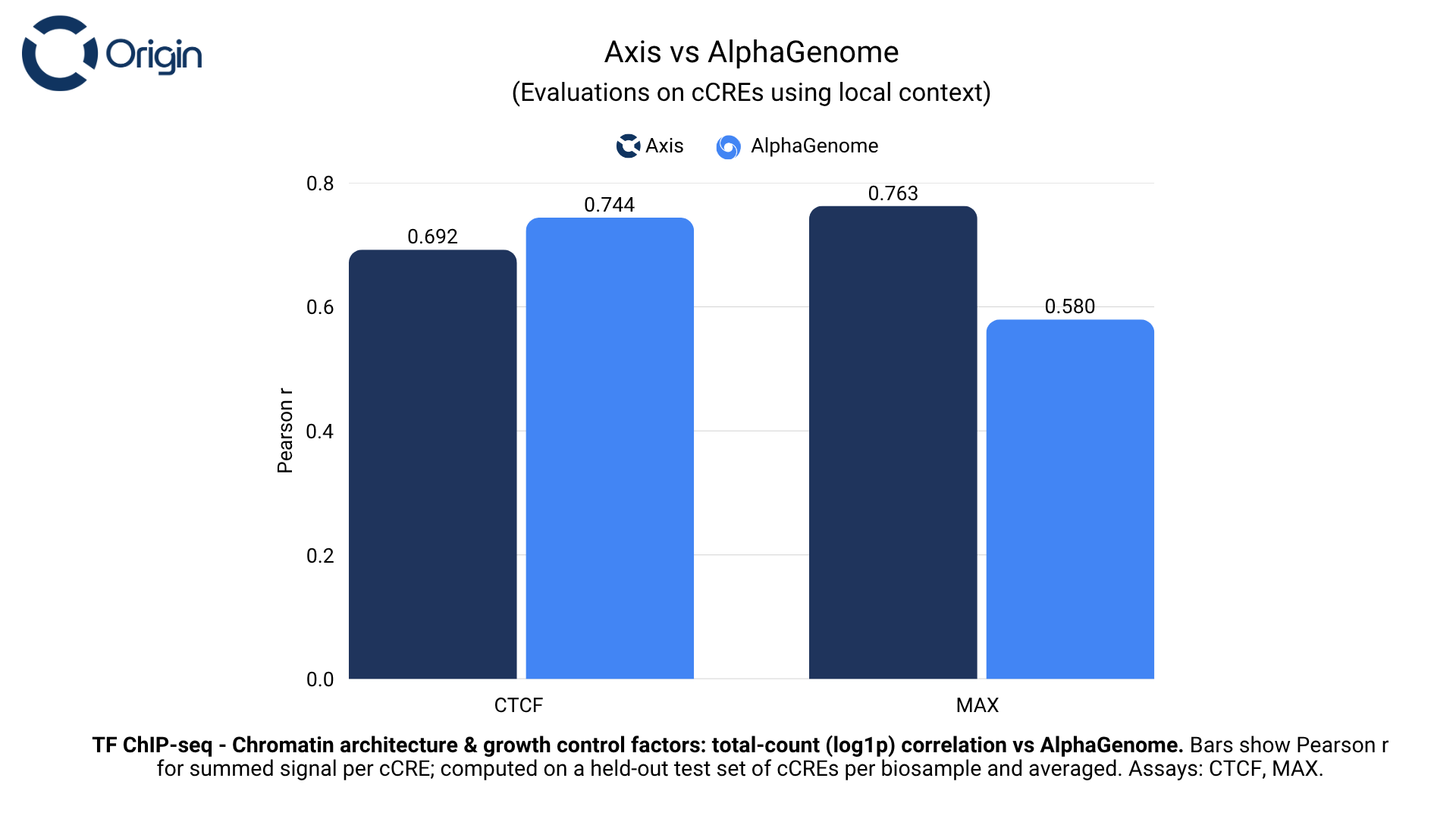
Chromatin architecture and growth control transcription factors perform functions like controlling structural access to regulatory elements and integrating growth signaling into transcriptional control.
Looking Ahead
Imagine having to use one ChatGPT for translating text and another for captioning images. This reality, where there were marginalized models for different tasks was not far away from materializing. A conscious effort to explore the benefits of multi-task learning eventually paved the way for today’s technology. One such work was “Multi-task sequence-to-sequence learning” by the Google Brain team. The work reported that training a model to caption images also improved translation quality between English and German. This would serve as one of the many precursors to the language models of today, with one of the paper’s authors, Ilya Sutskever, going on to create the first GPT.
The success of this paradigm in the natural language processing domain was an initial motivation behind our pursuit of a multifunctional DNA model. With Axis, we report that training a model to generate DNA sequences at a high resolution also improved the model’s ability to predict the function of existing DNA sequences.
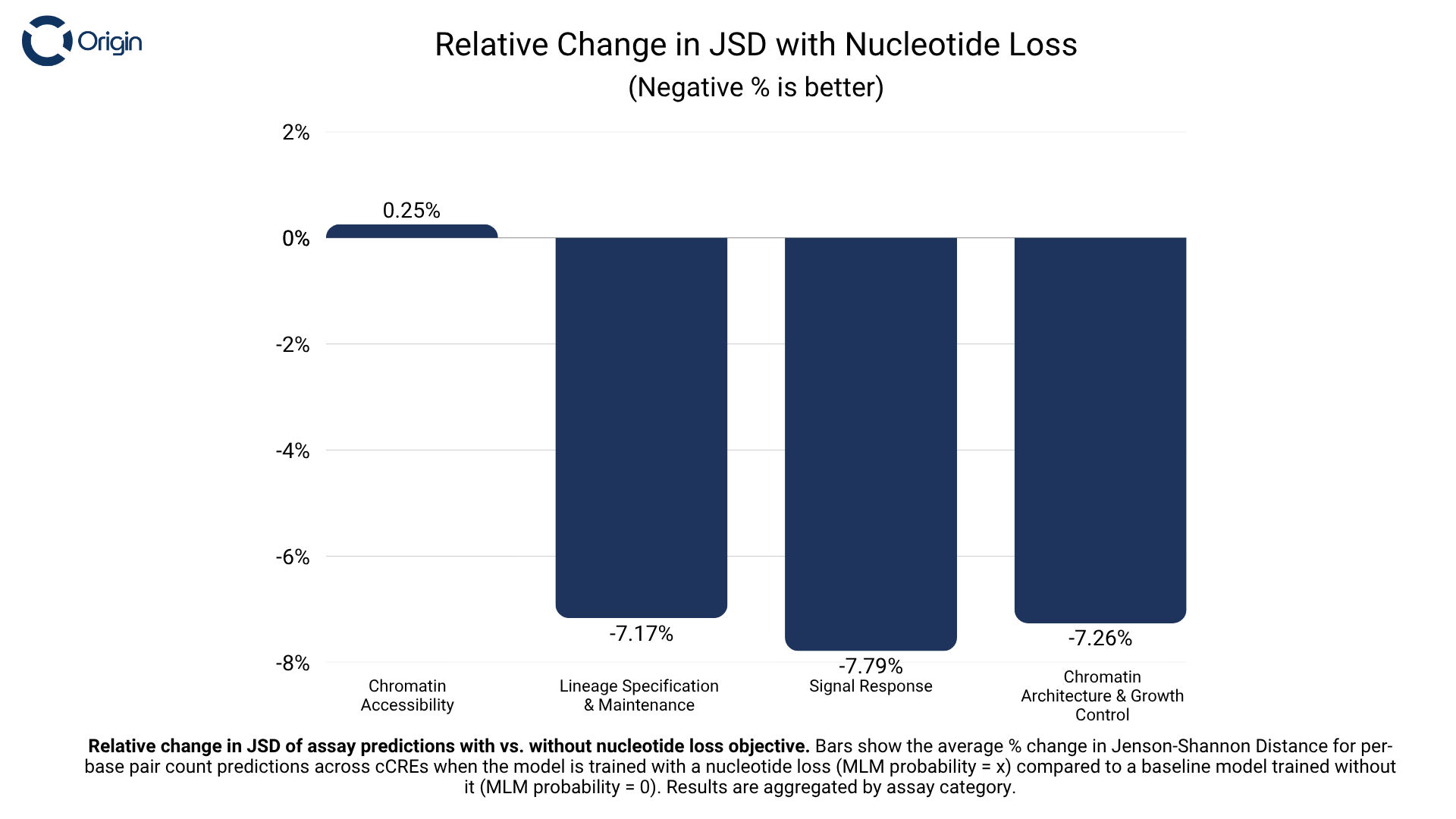
We’re excited about the implications of this and believe that Axis is just the tip of the iceberg when it comes to how promptable and unified models can and should be. Unified not just across functions within one domain like DNA, but across RNA, proteins and cellular representations. We believe a fundamental reason behind the lack of reliability and limited capabilities of today’s models is underspecification of the whole system. Biology is incredibly complex and in constant coordination with flow of information across modalities. We must make AI models that learn to compress and reason over the whole puzzle and not just pieces of it. Progress at every step of the way will make human lives better. Completing this grand challenge will unlock a new era of developing therapies that are capable of addressing highly complex diseases.
We are excited to take the model into a wet-lab and test out the model’s diverse capabilities. We’re interested in sharing access to the model via a free API to research institutions and interested individuals in order to get feedback. If you are interested in trying out the model, please fill out the form here.
If this work excites you and you want to dedicate your time and effort to tackling this problem with us, please reach out at hiring@origin.bio. We’re looking for exceptional engineers and researchers.